Istituto di Ricerche Farmacologiche Mario Negri IRCCS
Centro di Ricerche Cliniche per le Malattie Rare Aldo e Cele Daccò
Centro di Ricerche Cliniche per le Malattie Rare Aldo e Cele Daccò
Introduzione
Cause
Popolazione colpita
Altri tipi di SEU e patologie correlate (SEU tipica, SEU secondaria e PTT)
La sindrome emolitico uremica (SEU) è una malattia rara caratterizzata da rottura di globuli rossi (anemia emolitica), basso numero di piastrine (trombocitopenia) e insufficienza renale acuta. La causa sottostante è un'alterazione dei piccoli vasi sanguigni, con formazione di microtrombi fatti di piastrine, che chiudono i piccoli vasi del rene (microangiopatia trombotica) e che rompono i globuli rossi di passaggio.
Dobbiamo distinguere tra due diversi tipi di SEU, quella tipica e quella atipica. La SEU tipica è la forma più frequente (oltre il 90% dei casi) ed è solitamente preceduta da un grave episodio di gastroenterite causato da un'infezione da Escherichia Coli, produttore di potenti tossine chiamate Shiga tossine (STEC). In particolare il ceppo di E.Coli più frequentemente coinvolto in Europa e in Nord America è il 0157:H7.
Negli ultimi anni è stato invece dimostrato come la SEU atipica sia una malattia legata a un'alterata regolazione di una parte del sistema immunitario, il sistema del complemento, con una causa genetica riconoscibile nel 60% dei casi. Un'importante distinzione fra le due forme della malattia è che i bambini colpiti da un primo episodio di SEU tipica in genere hanno sintomi meno gravi, rispondono bene alla terapia e guariscono completamente, mentre i bambini con SEU atipica sono più facilmente esposti a importanti complicanze come l'insufficienza renale e l'ipertensione arteriosa severa. La sindrome emolitico uremica atipica può diventare una condizione cronica e i pazienti possono andar incontro a ripetute recidive nell'arco della loro vita.
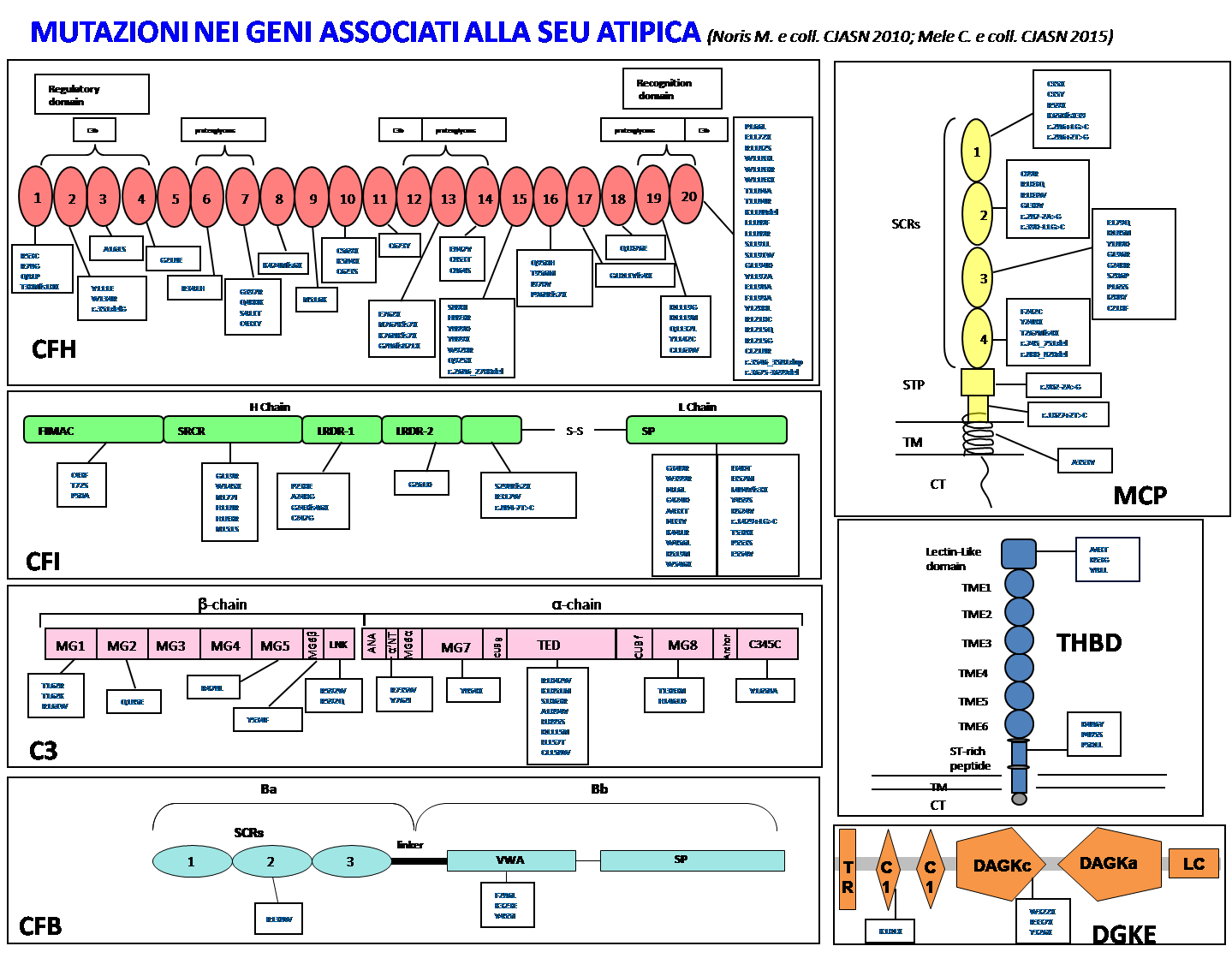
Recenti studi hanno dimostrato che il 60% circa dei casi di SEU atipica sono associati ad anomalie genetiche del sistema del complemento (in particolare della cosiddetta "via alternativa"), che è una parte fondamentale del sistema immunitario per la risposta alle infezioni. Il complemento è formato da una serie di proteine che, attivandosi, creano una cosiddetta "cascata", che porta all’eliminazione dell'agente infettivo, sia direttamente sia tramite altre cellule. Il complemento è finemente regolato in modo tale da prevenire danni alle cellule sane del proprio organismo. Questa delicata regolazione è basata su un certo numero di regolatori ancorati alla membrana (MCP, CR1,CR2, DAF, CD59 e CRIq) e fattori circolanti prodotti dal fegato (CFH, CFI, CFB, C3, C1-inh, C4BP), che proteggono i tessuti dell'ospite. I difetti genetici di alcune di queste proteine regolatrici del complemento causano una ridotta protezione della superficie endoteliale (quindi della parete dei vasi) contro l’attivazione del complemento.
In particolare, circa il 20-30% dei casi di SEU atipica sono associati ad anomalie del gene responsabile della produzione del fattore H (CFH), una proteina prodotta dal fegato che gioca un ruolo importante nella regolazione della via alternativa del complemento. Quando il fattore H non funziona in modo adeguato (a causa di un difetto genetico che determina perdita di funzione "loss-of-function"), si corre il rischio che il complemento si attivi in modo sregolato e che i vasi sanguigni del rene vengano danneggiati, causando le anomalie che portano alla formazione di trombi. Ad oggi, oltre 50 diverse mutazioni di CFH sono state identificate; la maggior parte sono in eterozigosi (ovvero presenti in una sola copia del gene) e sono raggruppate in una porzione particolare del gene (SCR 19-20). La quantità di fattore H nel sangue può essere dosata ma, anche se risulta nei limiti di norma, non si può escludere del tutto una mutazione nel gene.
Oltre alla mutazione del gene CFH ci può essere anche un meccanismo autoimmune sottostante: si possono formare cioè autoanticorpi contro il fattore H che ne alterano la funzione, descritti nel 6-10% dei casi di SEU atipica, soprattutto nell’età scolare. Il meccanismo con il quale questi autoanticorpi si formano non è ancora chiaro; c’è però una particolare "conformazione" genetica (delezione di CFHR1-3 in omozigosi, ossia presente in entrambe le copie dei geni) che si trova solitamente in associazione agli anticorpi contro il fattore H, ma che è presente anche nel 3-4% della popolazione sana, quindi non può essere considerata la causa della malattia.
Una rara causa di SEU atipica è inoltre rappresentata da riarrangiamenti tra geni collegati al fattore H (in particolare tra il gene CFH e il gene CFHR1), che determinano la formazione di un gene ibrido CFH/CFHR1 e, come conseguenza, un'anomalia di funzione del fattore H.
Altri casi di SEU sono associati a mutazioni genetiche tipo "loss-of-function" di altri geni che codificano per proteine regolatrici del sistema del complemento, come la proteina cofattore di membrana (MCP) ed il fattore I, che risultano alterati e mal funzionanti in presenza di una mutazione genetica. Nella SEU atipica sono state descritte anche mutazioni (che determinano aumento di funzione) in proteine chiave della via alternativa del complemento, quali fattore B (CFB) e C3, che risultano iperattive e quindi dannose. Inoltre, mutazioni nel gene che codifica per la trombomodulina (THBD), una glicoproteina anticoagulante della parete dei vasi con proprietà regolatrici del sistema del complemento, sono state scoperte nel 3-5% dei pazienti affetti da SEU atipica.
È stato inoltre evidenziato che circa il 3% dei pazienti ha due (o anche tre) mutazioni in diversi geni regolatori del complemento. Infine, recentemente sono state trovate mutazioni genetiche nel gene che codifica per diacilglicerol kinasi epsilon (DGKE), un enzima intracellulare che non sembra associato al sistema del complemento.
Clicca per ingrandire
L'identificazione di mutazioni in CFH, MCP, CFI, CFB, C3, THBD o DGKE ha implicazioni cliniche. Infatti, a seconda delle diverse mutazioni genetiche, si è visto che la SEU si può manifestare in modo molto diverso e può essere più o meno grave; per questo è importante fare un’analisi genetica completa per capire meglio la propria malattia e le prospettive per il futuro.
Prima dell'avvento di Eculizumab, circa il 60-70% dei pazienti con mutazioni in CFH, CFI e C3, e un terzo dei bambini con autoanticorpi contro il fattore H, perdevano la funzione renale o morivano durante il primo episodio, oppure sviluppavano insufficienza renale terminale in seguito a recidive. Anche mutazioni in CFB e THBD erano spesso associate a compromissione della funzione renale. Inoltre, circa il 20% dei pazienti con mutazioni in CFH avevano complicazioni cardiovascolari e un maggiore tasso di mortalità. La prognosi di questi malati è stata radicalmente modificata dall’avvento di eculizumab.
L'episodio acuto di SEU in pazienti con mutazioni in MCP è invece di per sé generalmente più lieve che in pazienti con mutazioni in CFH, o in uno degli altri geni del complemento, e l’80% di pazienti mutati in MCP va incontro a remissione completa. Le recidive sono frequenti, ma circa il 70-80% dei pazienti rimane a lungo senza necessità di dialisi. I pazienti con mutazioni nel gene DGKE hanno di solito un esordio molto precoce, entro il primo anno di vita, spesso hanno una perdita di proteine nelle urine (proteinuria) anche notevole (sindrome nefrosica), e tendono ad avere un decorso molto lento della malattia.
Per questi motivi genotipizzare i pazienti con SEU atipica può permettere un'ottimizzazione del trattamento.
Esistono rari casi di forme familiari di SEU atipica, in cui diversi membri di una stessa famiglia sono affetti dalla malattia: in questi casi si trova solitamente una causa genetica sottostante. Nella maggior parte delle famiglie con SEU atipica la malattia è trasmessa ai figli in modo autosomico dominante (50% di probabilità di trasmissione del difetto genetico dai genitori ai figli), mentre in un minor numero di famiglie (in particolare con mutazioni nel gene DGKE) la malattia è trasmessa con modalità autosomica recessiva (25% di probabilità di trasmissione del difetto genetico dai genitori ai figli). I maschi hanno la stessa probabilità delle femmine di ereditare la mutazione. La "penetranza" della mutazione genetica non è però completa: questo vuole dire che nella stessa famiglia ci possono essere persone che hanno la mutazione ma che non hanno la malattia, mentre altre con la stessa mutazione che sviluppano la SEU. Sembra inoltre che ci siano altri fattori legati ai geni del complemento (polimorfismi) che, combinati con le mutazioni, possano favorire l'esordio della malattia.
La SEU non è una patologia genetica classica ma una patologia complessa multifattoriale, dove per lo sviluppo della malattia sono importanti anche fattori cosiddetti ambientali, diversi per ogni persona. Infatti il primo episodio di SEU atipica è spesso preceduto da un'infezione batterica o virale, che si comporta da fattore scatenante in chi ha una predisposizione genetica. Anche la gravidanza, e in particolare il periodo post parto, rappresentano fattori scatenanti in donne geneticamente predisposte.
La SEU atipica è una condizione che raramente guarisce in modo completo, infatti, si possono avere recidive, soprattutto nei primi anni dopo l'episodio acuto, e molti pazienti hanno ricadute anche dopo un eventuale trapianto di rene. La possibilità che la malattia si ripresenti dopo il trapianto dipende anche da quale gene è implicato. Nei pazienti con SEU atipica in lista d'attesa per trapianto di rene andrebbe quindi eseguito uno screening completo di tutti i geni associati alla SEU, per permettere di valutare il rischio di ricorrenza della malattia.
La SEU atipica è una malattia molto rara e rappresenta meno del 5% di tutti i casi di sindrome emolitico uremica. La prevalenza è stimata essere di 1-9 casi per milione (www.orpha.net).
L'esordio della malattia è molto variabile, infatti, può avvenire nel periodo neonatale come nell’adulto, ma più spesso colpisce i bambini. Quando l’esordio è molto precoce è altamente probabile che ci sia una causa genetica sottostante.
La prognosi della malattia in passato era poco favorevole, con un'alta probabilità di insufficienza renale terminale con necessità di dialisi, e una mortalità che poteva arrivare fino al 10-15%. I recenti schemi terapeutici, che includono l'Eculizumab (un farmaco che inibisce il sistema del complemento), hanno permesso di ridurre notevolmente la percentuale di pazienti che necessitano di dialisi e di ridurre le complicanze.
La SEU tipica, associata all'infezione da Escherichia Coli produttore di tossina Shiga, è una malattia che colpisce in modo prevalente bambini tra 1 e 10 anni di vita, con un picco di incidenza attorno ai 3 anni. L'esordio della SEU tipica è preceduto da una gastroenterite caratterizzata da vomito, dolore addominale, febbre e diarrea spesso sanguinolenta. I sintomi della SEU tipica di solito si manifestano 10 giorni dopo lo sviluppo della gastroenterite e includono pallore, irritabilità, stanchezza, letargia ed eliminazione di minor quantità di urina. La malattia progredisce con l'inabilità dei reni ad eliminare le sostanze di scarto (insufficienza renale o uremia) e con l'insorgenza di anemia emolitica e di piastrinopenia. La formazione di trombi nei piccoli vasi (microangiopatia trombotica) può portare alla sofferenza di diversi organi oltre al rene, come il cervello. Talvolta, infatti, si possono osservare anche sintomi neurologici quali parestesie, crisi epilettiche, disorientamento e confusione fino al coma.
La causa più frequente di SEU tipica è l'infezione da Escherichia coli produttore di tossina Shiga, in particolare il ceppo 0157:H7.
Nell'epidemia del 2010 che ha colpito la Germania, causata dal ceppo di Escherichia Coli 0104:H4, la presentazione e l'evoluzione della malattia sono state inusuali. Circa il 90% dei pazienti, infatti, erano adulti e, in confronto alle precedenti epidemie, c'è stato un numero maggiore di donne giovani e di casi che hanno richiesto la dialisi (20% vs 6%); maggiore è stato anche il numero dei decessi (6% vs 1%).
La SEU può anche essere secondaria ad altre condizioni, quali:
La porpora trombotica trombocitopenia (PTT) o Sindrome di Moschowitz è una rara malattia del sangue anch'essa caratterizzata dalla presenza di trombi nei piccoli vasi arteriosi (microangiopatia trombotica). Esiste una grande somiglianza nella presentazione clinica, nei segni e sintomi, fra la PTT e la SEU. Tuttavia, esse sono condizioni che è importante distinguere e caratterizzare con rapidità perché devono essere gestite e trattate in modo diverso. La SEU colpisce più frequentemente i bambini, mentre la PTT colpisce tipicamente gli adulti e le donne intorno alla III-IV decade di vita. Per quanto le manifestazioni cliniche siano molto simili, nella PTT sono più frequenti i sintomi neurologici (parestesie, crisi epilettiche, cefalea, confusione mentale e coma) mentre nella SEU prevale l'interessamento renale. La PTT può essere dovuta alla carenza congenita di un enzima che regola la coagulazione del sangue (ADAMTS13) o, più spesso, alla presenza di anticorpi contro questo enzima (meccanismo autoimmune). È quindi importante nelle prime fasi effettuare un dosaggio di questo enzima e una ricerca di eventuali auto-anticorpi.

